All data submissions start with completing an intake form via the AD + EL Service Desk. A complete and thorough intake supports the FAIR principles (Findable, Accessible, Interoperable, and Reusable) and helps our team understand the data being shared and how/when they will be released.
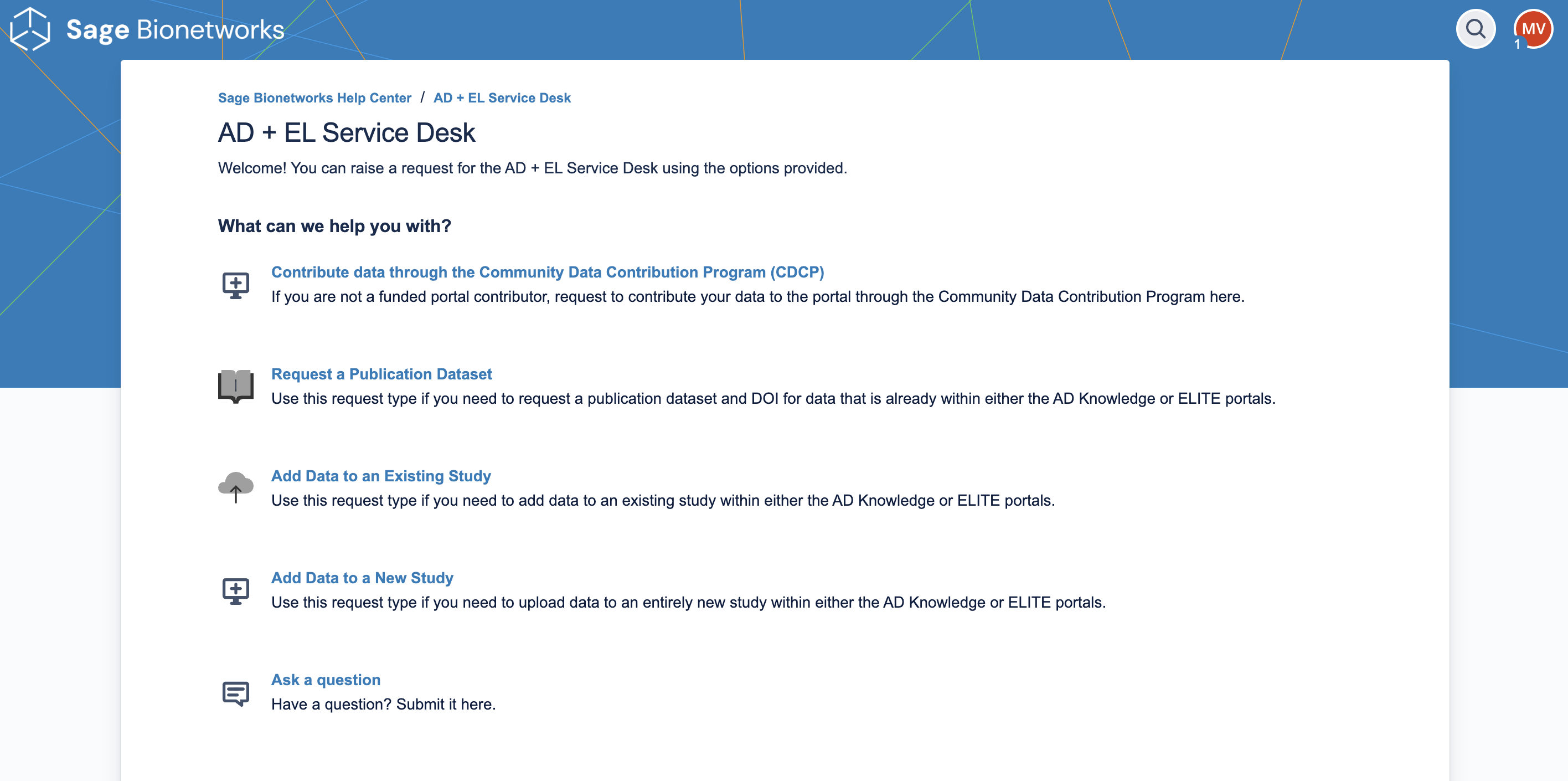
Add Data to a New Study: Use this request type if you need to upload data to an entirely new study within either the AD Knowledge or ELITE Portals.
Add Data to an Existing Study: Use this request type if you need to add data to an existing study within either the AD Knowledge or ELITE Portals.
Request a Publication Dataset: Use this request type if you need to request a publication dataset and DOI for data that is already within either the AD Knowledge or ELITE Portals.
Contribute data through the CDCP: The Community Data Contribution Program (CDCP) is a means for sharing data and other resources that are not directly funded through the National Institute on Aging (NIA) at the National Institutes of Health (NIH). Data submissions through the CDCP will be reviewed by the EL DMCC Steering Committee for scientific relevance to the ELITE Portal.
Ask a question: Use this request type to submit a question to the EL DMCC team.
In the top right corner, you can also access a log of all AD + EL Service Desk tickets you have created and/or are assigned to. The AD + EL Service Desk is powered by Jira Service Management.
How to Create an AD + EL Service Desk Request
1. Select the request type matching your situation
(for first-time data contributors) Select “Add Data to a New Study”
(for returning data contributors) Select “Add Data to an Existing Study”
(for manuscript authors needing a DOI for data residing in the ELITE Portal) Select “Request a DOI for Publication”
2. Complete the required form fields and submit the form
Once a form is submitted, a Service Desk ticket will be opened on your behalf. This ticket is used to track your request and communicate with your team regarding any questions or issues about the submission.
Information you will need to submit a request
The following information will be requested when you Submit a Service Desk Request with us and complete intake for your submission. Please be prepared to input this information into the Service Desk form.
Project information
Related Service Desk form questions
Program name
Project name
Grant Number
Originating funding source
What is the name of the institution(s) where the data was generated?
What is the name of the institution(s) where biospecimens were collected?
Please provide the name for the project and grant number that your submission is affiliated with. A list of Exceptional Longevity projects can be found here.
Team roles
Related Service Desk form questions
Grant PI Name
Institution of Lead PI
Data Liaison Name and Email
Data Uploader name and Email
Each data contributing team should assign people in these roles:
Role
Description
Responsibilities
Grant PI
This person authorizes the activity of the study (also may be called team lead).
Send signed DTA (if required) and sign the data attestation form
This person is responsible for assembling the required metadata and uploading the data and metadata to Synapse, and therefore, they must be familiar with R, Python, or the Command Line in order to use the Synapse API clients
Become a registered and certified Synapse user
Assemble metadata
Upload data files and annotate data through the data curator app (DCA)
*If appropriate, the data liaison may also be the data uploader.
Data upload details
Related Service Desk form questions
Will the data be indexed?
If Yes to Data Index, please provide a stable URL for the dataset(s)
Data type(s)
Data upload details
Does this contribution contain human data?
Did the human subjects consent to data sharing in a repository? (Y/N)
Specific data use conditions for this data
Please provide details about the data being uploaded.
A data indexis used when the data is publicly and permanently hosted elsewhere through an already established link. All data contribution steps will be completed except uploading the data to Synapse. We will instead point to the data from the ELITE portal and use our metadata templates to provide data discoverability.
The data contributor is responsible for providing a stable URL.
Requests to index data must be approved by the NIA and require a justification for why the data must be deposited into another repository outside of Synapse.
To submit a request, email elite-portal@sagebase.org. Please provide a summary of the data to be indexed, the repository where the data will be hosted, and a justification for why this data should reside there.
Study description
Related form questions
Unique study name and abbreviation
Study description
A key data organizing unit in the portal is the Study. Associated with a Study is:
a description of the cohort or model system the data has or will be generated from
summaries of the methods used to generate the data (think about this as the similar type of information you would write for a well written Materials and Methods section in a paper)
the data itself
associated metadata
an acknowledgment statement that users of the data will be requested to put in manuscripts and presentations
Data generated through a grant may be grouped into one or multiple studies. The EL DMCC will work with you to determine if data from your grant should be grouped into one or multiple studies based on the information in the data contribution survey.
A complete study description provides a summary of the cohort or model system your data comes from and includes:
A Descriptive Study Name: This should be a short descriptive sentence.
Example - “The Mount Sinai Brain Bank study”.
A Study Name Abbreviation: This should be an abbreviation of the descriptive study name and will be used to annotate all content belonging to a specific study.
Example - “MSBB”.
A Study Description: Describe the human cohort(s) or model system(s) the data comes from. Add links or references for additional information if available. Include additional details relevant to the specified cohort or model system, as detailed in the examples below.
Study Description example: Human cohort
⚠️ Include the study type (randomized controlled study, prospective observational study, case-control study, or post-mortem study), disease focus, diagnostic criteria, and inclusion/exclusion criteria of study participants. For post-mortem studies include the tissue bank name(s).
Brain specimens were obtained from the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB) which holds over 1,700 samples. This cohort was assembled after applying stringent inclusion/exclusion criteria and represents the full spectrum of disease severity. Neuropathological assessments are performed according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) protocol and include assessment by hematoxylin and eosin, modified Bielschowski, modified thioflavin S, and anti-β amyloid (4G8), anti-tau (AD2) and anti-ubiquitin (Daka Corp.). Each case is assigned a Braak AD-staging score for progression of neurofibrillary neuropathology. Quantitative data regarding the density of neuritic plaques in the middle frontal gyrus, orbital frontal cortex, superior temporal gyrus, inferior parietal cortex and calcarine cortex are also collected as described . Clinical dementia rating scale (CDR) and mini–mental state examination (MMSE) severity tests are conducted for assessment of dementia and cognitive status. Final diagnoses and CDR scores are conferred by consensus. Based on CDR classification , subjects are grouped as no cognitive deficits (CDR = 0), questionable dementia (CDR = 0.5), mild dementia (CDR = 1.0), moderate dementia (CDR = 2.0), and severe to terminal dementia (CDR = 3.0–5.0). Covariates including demographic and neuropathological data were collected on the samples used for this project including postmortem interval, race, age of death, clinical dementia rating, clinical neuropathology diagnosis, CERAD, Braak, sex, and a series of neuropathological variables. See the Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer's disease for a detailed description of the study and the data.
Study Description example: Animal model
⚠️ Include species, treatments, (if genetically modified) genotype and genetic background. Provide a link to the strain datasheet(s) if a commercial model, or a description of how it was created if not.
This is a study of a familial Alzheimer's disease model expressing mutant human APP and PS1. The 5XFAD transgenic mice overexpress both mutant human APP(695) with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) Familial Alzheimer's Disease (FAD) mutations and human PS1 harboring two FAD mutations, M146L and L286V. Expression of both transgenes is regulated by neural-specific elements of the mouse Thy1 promoter to drive overexpression in the brain. These 5XFAD transgenic mice rapidly recapitulate major features of Alzheimer's Disease amyloid pathology and may be useful models of intraneuronal Aβ-42 induced neurodegeneration and amyloid plaque formation. See more information on this mouse model through the Experimental Tools.
Study Description example: In-vitro cell culture
⚠️ Include species, cell type, cell culture information (such as primary or immortalized cell line, passage, treatments, differentiation). If a commercial cell line, provide a link.
For iPSCs and organoids, include the following:
Details of the reprogramming protocol, including transcription factors or transgenes, any commercial kits used, and any cell quality control checks for pluripotency, genomic integrity, or other markers
Details of the differentiation protocol, including any intermediate cell types (i.e., NPCs or NSCs), growth factors, induction transgenes, culture media (specify feeder-dependent or feeder-free), plate coating or matrix
Any additional treatments or genetic alterations
Note: reprogramming, differentiation, and culture protocols may be provided as additional documents with your data submission.
This study is a comparison of human pluripotent stem cell derived microglia to human primary microglia, human primary hepatic macrophages and peripheral blood derived macrophages non-polarized or polarized with IFN-γ and LPS, IL-4 and IL-13 or IL-10.
Microglia, the immune cells of the brain, are crucial to proper development and maintenance of the central nervous system, and their involvement in numerous neurological disorders is increasingly being recognized. To improve our understanding of human microglial biology, we devised a chemically defined protocol to generate human microglia from pluripotent stem cells. Myeloid progenitors expressing CD14/CX3CR1 were generated within 30 days of differentiation from both embryonic and induced pluripotent stem cells (iPSCs). Further differentiation of the progenitors resulted in ramified microglia with highly motile processes, expressing typical microglial markers. Analyses of gene expression and cytokine release showed close similarities between iPSC-derived (iPSC-MG) and human primary microglia as well as clear distinctions from macrophages. iPSC-MG were able to phagocytose and responded to ADP by producing intracellular Ca2+ transients, whereas macrophages lacked such response. The differentiation protocol was highly reproducible across several pluripotent stem cell (PSC) lines.
Cell culture: As microglia cells arise from myeloid progenitors in the yolk sack during embryonic development, we established a serum- and feeder-free protocol to differentiate human PSCs towards the myeloid lineage.
Growth Protocols
iPSC-MG(iPSC1-derived microglia): 4 days mTeSR Custom medium with 80ng/ml BMP4. 2 days in STEM-Pro34 with 2mM GutaMAX-I, 25ng/ml bFGF, 100ng/ml SCF and 80ng/ml VEGF. 8 days in StemPro-34 containing 50ng/ml SCF, 50ng/ml IL-3, 5ng/ml TPO, 50ng/ml M-CSF and 50ng/ml Flt3 ligand. 10-20 days in StemPro-34 containing 50ng/ml M-CSF, 50ng/ml Flt3 ligand and 25ng/ml GM-CSF until CD14+ and/or CX3CR1+ progenitors were isolated. Finally cells were cultured in RPMI-1640 supplemented with 2mM GlutaMAX-I, 10ng/ml GM-CSF and 100ng/ml IL-34 for 2 weeks.
PB-M(-) (Non-polarized macrophages): CD14+ cells isolated from peripheral blood were cultured for 7 days in ultra-low attachment in RPMI-1640 with 2mM GlutaMAX-I, 10% heat-inactivated human serum and 20ng/ml M-CSF.
hMG (Human primary microlgia): Primary microglia were cultured in Microglial Medium (ScienCell)
PB-M(LPS,INFγ) (Macrophages polarized with IFN-γ and LPS), PB-M(IL4, IL13) (Macrophages polarized with IL-4 and IL-13), and PB-M(IL10) (Macrophages polarized with IL-10): CD14+ cells isolated from peripheral blood were cultured for 5 days in ultra-low attachment in RPMI-1640 with 2mM GlutaMAX-I, 10% heat-inactivated human serum and 20ng/ml M-CSF followed by 2 days in the same medium plus the cytokine(s) indicated in the brackets.
Methods description
Related form questions
Methods description
For each assay or assessment, provide a summary of:
sample processing
data generation
data processing, including which organs and tissues the samples came from.
For other tests (such as cognitive assessments or imaging), include a description of how the test was done.
Include links to any commercial equipment or tools, or code repositories. Detailed protocols are highly recommended. These can be uploaded as pdf together with the data-files, or as links to protocol repositories such as http://protocols.io or Open Lab Notebooks.
(link to additional information in the text, or add a list of references)
Example: RNA sequencing
Sample processing: This distribution contain samples isolated from from Brodmann Areas 10, 22, 36 and 44. The specific brain regions were dissected while frozen from flash frozen never-thawed ~8 mm thick coronal tissue blocks using a dry ice cooled reciprocating saw. The dissected regions were then pulverized to a fine powder consistency in liquid nitrogen cooled mortar and pestle and distributed into 50 mg aliquots. All aliquots were barcoded and stored at -80oC until RNA isolation. RNA samples were isolated at two RNA preparation cores in Mt. Sinai, HIMC and qPCR. At both cores, the total RNA were isolated from brain tissues using RNeasy Lipid Tissue Mini Kit from Qiagen (cat#74804) according to the manufacturer's protocol (The RNeasy Lipid Tissue Mini Kit Handbook, Qiagen 104945, 02/2009) with slight modifications.
Library preparation :Preparation of samples for RNA-Seq analysis was performed using the TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, CA). Briefly, rRNA was depleted from total RNA using the Ribo-Zero rRNA Removal Kit (Human/Mouse/Rat) (Illumina, San Diego, CA) to enrich for coding RNA and long non-coding RNA. The cDNA was synthesized using random hexamers, end-repaired and ligated with appropriate adaptors for sequencing. The library then underwent size selection and purification using AMPure XP beads (Beckman Coulter, Brea, CA). The appropriate Illumina recommended 6-bp bar-code bases are introduced at one end of the adaptors during PCR amplification step. The size and concentration of the RNAseq libraries was measured by Bioanalyzer (Agilent, Santa Clara, CA) and Qubit fluorometry (Life Technologies, Grand Island, NY) before loading onto the sequencer.
Sequencing : The Ribo-Zero libraries were sequenced on the Illumina HiSeq 2500 System with 100 nucleotide single end reads, according to the standard manufacturer’s protocol (Illumina, San Diego, CA). The raw sequence reads were aligned to human genome hg19 with the star aligner (v2.3.0e). Then the gene level expression (read counts) were quantified by featureCounts (v1.4.4) from the Subread package. Genes with at least 1 read count in at least 10 libraries were considered present, otherwise removed. The trimmed mean of M-values (TMM) normalization method in the R/bioconductor edgeR package was employed to estimate scaling factors so as to adjust for differences in library sizes. The data was corrected for known covariates factors, including PMI, RACE, Batch, SEX, RIN and Exonic rate to remove the confounding effects.
Alignment and quantification : The raw sequence reads were aligned to human genome hg19 with the star aligner (v2.3.0e) and gene level expression (read counts) were quantified by featureCounts (v1.4.4) from the Subread package.
Normalization and covariates correction : Genes with least 1 read count in at least 10 libraries were considered present, otherwise removed. The trimmed mean of M-values (TMM) normalization method in the R/bioconductor edgeR package was employed to estimate scaling factors so as to adjust for differences in library sizes. Known covariate factors, including batch, sex, race, age, RIN, PMI, exonic rate and rRNA rate were corrected using a linear model to remove the confounding effects.
Sample filter : Following the QC described below, samples with QC actions “Remap” or “Exclude”, low RIN score (<4), or relatively large rRNA rate (>5%) were removed.
Quality Control : Sample quality control (QC) was performed through the cross-data type genetic similarity analysis by making use of genetic variants identified in different types of sequencing data (WGS, WES and RNA-seq) in the MSBB AD cohort. Briefly. we estimated pairwise sample kinship using KING and compared the genetic concordance among all sequencing samples across different data types. Since the sequencing samples from the same brain are expected to have high genetic similarity (in theory identical) while the sequencing samples from different brains are expected to present low genetic similarity, any kinship failure or mismatch suggested sample errors due to either incorrect ID labeling, sample swapping or contamination. We flagged the suspicious sample pairs which should match genetically but did not, and spurious sample pairs which should not match but present very high genetic similarity. Using an iterative procedure coupled with a majority voting scheme, we sequentially tested whether every suspicious or spurious sample was mislabeled and could be unambiguously identified with the correct brain source. The QC procedure is detailed in Wang et al , and results listing remapped or removes samples listed in the MSBB_RNAseq_covariates assay metadata file.
Example: Mouse behavioral assessment
All behavioral assays were performed on mice (4, 6, 12 months) generated and housed at The Jackson Laboratory.
Housing conditions : Mice were bred and aged in the main Research Animal Facility prior to testing in the Neurobehavioral Phenotyping Facility (NPF) procedure rooms located in adjacent connected buildings, including the Center for Biometric Analysis (CBA). The dedicated NPF housing room consists of PIV caging with temperature controlled at a setting of 72±2°F and humidity at 50±20%. The testing facility was on a 12:12 L:D schedule (lights on at 6:00 am) with all testing performed during the light cycle (typically with testing beginning 1 hour after lights on and concluding 1 hour before lights off, with the exception of wheel running which was continuous 24-hour testing for up to 5 days). At minimum 7 days prior to the start of behavioral testing, subjects were individually housed at 2.5, 4.5, and 10.5 months of age - approximately 45 days prior to tissue harvest endpoint, allowing sufficient time for all behavioral testing. All subjects were randomized and counterbalanced for testing order across multiples of instrumentation and time of day for each test day, with a simplified testing ID number (e.g. #1-100), and all technicians were blinded to genotype which was coded (e.g. A, B, C, etc.). The blind was maintained throughout testing and until after the data were analyzed.
Testing Battery Order : Behavioral tests were conducted as previously reported (Sukoff Rizzo et al 2018 Current Protocols in Mouse Biology) in the following order with at minimum a 1-2 day rest period between tests: frailty assessment with core body temperature recording, open field test, spontaneous alternation, rotarod, episodic memory, novel spatial recognition short term memory task, and wheel running. On each test day, subjects were transported from the adjacent housing room into the procedure room, tails were labeled with a non-toxic permanent marker with the assigned subject ID number, and subjects were left to acclimate to the testing environment for a minimum 60 minutes prior to testing. Between subjects, all testing arenas were sanitized with 70% ethanol solution and dried prior to introducing the next subject. Lighting in the testing rooms were consistent with the housing room (~ 500 lux) unless where specifically noted. At minimum 5 days post the conclusion of behavioral testing, mice were sent for tissue harvesting per DMP protocols.
Frailty Assessment : The frailty assessment was conducted as previously published (Sukoff Rizzo et al Current Protocols Mouse Biology 2018) and is used to assess the presence of a spectrum of aging related characteristics in mice. Briefly, test subjects were transported to the testing room, body weights were recorded and tails were labeled with the subject ID number (e.g. 1-50) using a non-toxic marker. Mice were then left undisturbed for a minimum of 60 min to acclimate to the procedure room. Following acclimation, subjects were individually evaluated for the absence or presence of 26 characteristic traits and reflexes and scored a 0, 0.5, or 1 (based on presence/absence, and severity) for each assessment by a trained observer, blind to genotype/age, and included the following assessments: alopecia; loss of fur color; dermatitis/skin lesions; loss of whiskers; coat condition; piloerection; cataracts; eye discharge/swelling; microphthalmia; nasal discharge; rectal prolapse; vaginal/uterine/penile; diarrhea; vestibular disturbance; vision loss assessed by visual placing upon subject being lowered to a grid; menace reflex; tail stiffening; impaired gait during free walking; tremor; tumors; distended abdomen; kyphosis; body condition; breathing rate/depth; malocclusions; righting reflex. The frailty index score was calculated as the cumulative score of all measures with a maximum score of 26.
Core Body Temperature : Core body temperature was recorded just prior to the conclusion of the frailty assessment via a glycerol lubricated thermistor rectal probe (Braintree Scientific product# RET 3; measuring 3/4" L .028 dia. .065 tip) inserted ~ 2cm into the rectum of a manually restrained mouse for approximately 10 sec. Temperature was recorded to the nearest 0.1°C (Braintree Scientific product#TH5 Thermalert digital thermometer).
Open field test : Versamax Open Field Arenas (40 cm x 40 cm x 40 cm; Omnitech Electronics, OH USA) were used for this test. Arenas were housed within sound attenuated chambers with lighting in the testing room and arenas consistent with the housing room (~500 lux). Mice were placed individually into the center of the arena and infrared beams recorded distance traveled (cm), vertical activity, and perimeter/center time. Data were collected in 5 min timebins for a duration of 60 minutes.
Data preprocessing : The RNAseq raw read counts data was normalized by applying the trimmed mean of M-values (TMM) method as implemented in R/bioconductor package edgeR to adjust for differences in library size. Covariate factors, including sex, race, age, RIN, PMI, batch and exonic rate, were corrected using a linear model to remove the confounding effects.
Coexpression networks construction : Coexpression networks were constructed for three brain regions (BM10, BM22 and BM36) from the normalized and covariates adjusted RNAseq data by using WGCNA and MEGENA
WGCNA : Networks were constructed by an R package of coexpp , an optimized version of the WGCNA package, with the parameter power determined by a scale free index > 0.8 and other settings by default. Note that these networks are built on a pre-QC version of the gene expression data.
MEGENA : Pearson correlation coefficients (PCCs) were firstly computed for all gene pairs. Significant PCCs at permutation based FDR 0.05 were ranked and iteratively tested for planarity to grow a Planar Filtered Network (PFN) from the PMFG algorithm. Multiscale Clustering Analysis (MCA) was conducted with the resulting PFN to identify coexpression modules at different network scale topology. Note that there are two version of the MEGENA networks. The latest version was built on the most current post-QC version of the expression data (see file provenance).
WINA : WINA is a R package which implements a computationally optimized procedure for Weighted Gene Coexpression Network Analysis (WGCNA). In WINA analysis, Pearson’s correlation coefficients were calculated between all pairs of genes. Next, the correlation matrix was converted into an adjacency matrix using a power function f(x) = x^β, where x is the element of the correlation matrix and parameter β was determined such that the resulting adjacency matrix was approximately scale-free. In the present study, we used β = 6 with other parameters set by default. The adjacency matrix was subsequently transformed into a topological overlap matrix (TOM) which captured both the direct and indirect interactions between a pair of genes. Average linkage hierarchical clustering was then employed to cluster genes based on the TOM. Finally a tree cutting algorithm was used to dynamically cut the hierarchical clustering dendrogram branches into highly connected modules, each of which was assigned a distinct color code.
Acknowledgement statement
Related form questions
Acknowledgement statement
The acknowledgement statement should include:
The Study Name Abbreviation
The following language (word-for-word): The results published here are in whole or in part based on data obtained from the ELITE Portal (http://eliteportal.synapse.org )
An additional paragraph acknowledging grants and/or people of your choice
Example Acknowledgement Statement
Study Name Abbreviation: MRGWAS
Statement: The results published here are in whole or in part based on data obtained from the ELITE Portal (https://eliteportal.synapse.org/ ). Data analysis was supported through funding by NIA grant U24AG051129. Published genome-wide association study (GWAS) results from Sebastiani et al., 2017 (PMID: 28329165) were kindly provided by Dr. Paola Sebastiani. All other GWAS results are publicly available through the GWAS catalog or MR-Base.